PEG in Lipid Nanoparticle (LNP) Formulations: Addressing Challenges and Seeking Solutions
- Michael Nguyen
- Mar 18, 2024
- 8 min read

Updated: Mar 29, 2024
Background
Polyethylene glycol (PEG) is a hydrophilic polymer unit which can be viewed in Fig. 1. It is dissolvable in common solvents, such as water and ethanol, making it a suitable modification and/or component for pharmaceutical applications [1]. PEG imparts a stealth-like quality to the conjugated molecule or formulation, increasing blood circulation half-life [2]. This mitigates immune system clearance and allows for the systemic or targeted distribution of these PEG-conjugated molecules or PEG-doped particles and improved sustained drug delivery [3].

Figure 1. Structure of PEG and two commonly used PEGylated lipids in lipid nanoparticle formulations. ALC-0159 and DMG-PEG(2000) are used in the Pfizer-BioNTech and Moderna COVID-19 vaccines, respectively.
PEG lipids gained worldwide prominence during the SARS-CoV-2 global pandemic. They were a critical part in the 4 lipid component formulations of the FDA approved Pfizer-BioNTech BNT162b2 and Moderna mRNA-1273 SARS-CoV-2 vaccines. Since their introduction, PEG and PEG-like variants have been at the forefront of new research and development efforts for a variety of therapeutic modalities. Despite this, there are glaring disadvantages to the continued use of PEGylated components, which we at Helix Biotech, Inc. will further delve into below.
The Prominence of PEG
The prominence of PEG and PEGylated materials is due to their long history of use in commercial and pharmaceutical applications. PEG was first synthesized in the 19th century, and would eventually be conjugated to therapeutic proteins for immunological protection by the 1970s [4]. This would pave the way for the first FDA approved PEGylated enzyme, Adagen, in 1990. At around the same period, researchers began incorporating PEGylated lipids into liposomes, resulting in the first FDA approved PEGylated liposome, Doxil, in 1995. Due to its many clinical successes, commercial availability, and extensive publicly available testing data, PEG was and still is used in therapeutic formulations today. To put it simply, it has a much longer history and greater public acceptance than its alternatives.
The PEG Effects on Particle Properties and Lifetime
PEG influences the various stages of the LNP pathway. Beginning with the formulation step, the incorporation of PEG helps to prevent LNP aggregation, stabilizing the nanoparticle structure, and promoting in vivo circulation time. Although the mole percent is low (between 1-5%), adjusting this percentage has been shown to modulate particle size and polydispersity [5].
Though PEGylated liposomes and LNPs increase circulation time of their cargo, it has been shown that PEG can actually hinder particle uptake into cells. For example, liposomal uptake via endocytosis was reduced by 5 fold or greater when compared to non-PEGylated liposomes [6]. This is likely due to the high percentage (5 mol %) of PEG, which may hinder the crucial PEG shedding stage required for efficient cellular uptake. Attaching a cell-penetrating peptide rescued and even enhanced uptake [6], demonstrating the potential of other technologies to work in parallel with PEG.
After the particles are taken up by cellular endocytosis, they and their drug content must be released through a mechanism dubbed “endosomal escape.” This is theorized to occur through an interplay with the other lipid components, leading to endosomal membrane rupture. But with improper PEG shedding, this step can be hindered, thereby reducing transfection rates [7].
PEG Immunogenicity
The first report of PEG inducing anti-PEG antibody production and immunological responses appeared in 1983 [8]. This finding would spur concerns about PEG’s potential and widespread use. Currently, the ongoing dilemma facing PEGylated medications is the “repeat dosing” issue. The COVID-19 mRNA-LNP vaccines require additional doses to boost waning anti-spike protein levels within the body. However, the repeat doses show an increase in local and systemic reactogenicity in humans [9]. This is presumably due to an increase in the adaptive immune response to the LNP, which leading theories suggest is related to the PEG component of the formulation [10].
Several notable studies show the BNT162b2 and mRNA-1273 vaccines can promote the production of anti-PEG antibodies in humans even after a single injection [11-14]. Flow cytometry on human plasma before and after PEG-LNP administration showed a clear correlation between LNP-immune cell association with pre-existing anti-PEG levels [12]. This leads the way for anti-PEG and PEG-LNP binding and activation of the complement system and the eventual phagocytosis and clearance of the LNP. Interestingly, these studies consistently show that Moderna’s mRNA-1273 causes a greater anti-PEG response than Pfizer’s BNT162b2. It is difficult to tell if this is due to the different nature of the PEG lipids employed, the higher mRNA and PEG content in the mRNA-1273 vaccine or some other factor(s) entirely. Despite these findings, it is difficult to correlate anti-PEG antibody levels with vaccine efficacy as anti-Spike protein IgG levels still rise sharply after the 2nd and additional booster doses [15,16].
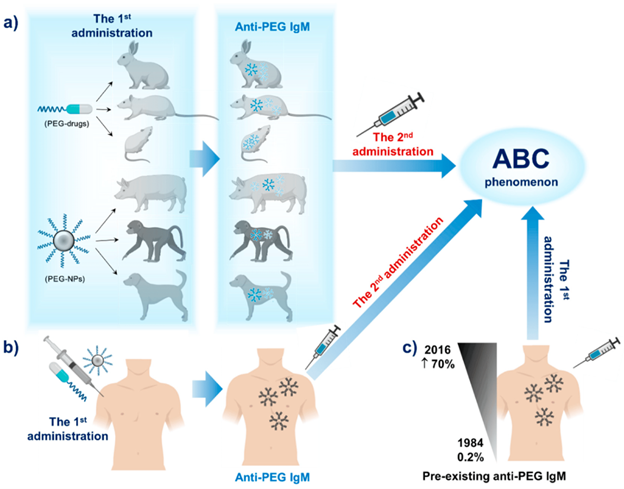
Figure 2. Illustration of the effects of repeated administration of PEGylated therapeutics in several (a) animal models and (b) humans. The 1st administration induces anti-PEG IgM production, while the 2nd causes the ABC phenomenon to occur. c) Pre-existing anti-PEG IgM from previous exposures results in ABC after just the 1st administration.
Credit: Hoang et al., 2019 [24].
Also, due to the increase in anti-PEG antibodies, these LNPs are susceptible to accelerated blood clearance (ABC). The ABC is thought to occur through the complement system, consisting of a complex array of proteins that work together to recognize, opsonize (mark for clearance) and destroy foreign pathogens, which can contribute to inflammatory conditions. These details would help explain the increase in local and systemic reactogenicity exhibited by human patients after additional vaccine doses [17]. Due to pre-existing anti-PEG antibodies, LNPs introduced via a second injection are taken up by Kupffer cells in the liver at a faster rate than found with the initial dose [18], reducing their interactions with target cells [19]. For a visual representation of how PEG administration and anti-PEG IgMs lead to ABC, refer to Fig. 2.
De-PEGylation and PEG Alternatives
Just as all other biomaterials, LNPs interact with their environment and one notable event is PEG shedding or de-PEGylation, i.e. the desorption of PEG from the LNP surface, and its replacement with a protein corona [20]. The protein corona is critical for LNP stability and even targeting of specific tissue by altering the LNP surface properties [21]. Dams et al. and Ishida et al. found that PEG clearance via the immune complement system occurred within minutes after injection - much faster than the typical PEG shedding mechanism that occurs on the order of hours [19].
To circumvent this would require a faster PEG shedding rate. This rate was shown to be dependent on the PEG length (shorter increases shedding) as shown by NMR [19]. Increasing shedding rate also decreased anti-PEG IgM generation [19]. The reactogenicity of the PEG component also relies on its terminal moiety, with methoxy-PEG exhibiting greater immunogenicity than hydroxy- and t-butoxy-PEG [22]. Mima et al. showed free PEG does not significantly increase anti-PEG antibody production as when they are incorporated into liposomes (and perhaps lipid nanoparticles). These results suggest there is an interplay between the PEG shedding rate and antibody generation. We must also keep in mind that a fast shedding is not necessarily a desired property as it removes away the stealth-like property conferred to the LNP by PEG.
Promising PEG technologies have been developed to initiate PEG shedding upon introduction to an acidic environment, such as the proximity of an endosome or tumor [7,23]. This has been shown to promote uptake and transfection rates but has not yet been proven in the clinic. There also exist many initiatives to replace PEG in LNP formulations, and for a more detailed review on these see here [24]. However, some of their issues mirror that of PEG - the production of polymer-specific antibodies.
Concluding remarks
PEGylated lipids serve the purpose of stabilizing LNPs throughout formulation, storage, and circulation. The PEG moiety introduces unfavorable steric interactions between particles, which prevents aggregation; this same property, however, inhibits LNPs from interacting with host cells, leading to reductions in cellular uptake and drug delivery. As a result, there is a fine balance between PEG shedding, the immunogenic response and the delivery of the drug payload. Still, the immunogenicity and reactogenicity of PEG and its alternatives must be further elucidated and overcome due to the repeat dosing need of necessary therapeutics for various maladies.
References
Brady, J., Dürig, T., Lee, P. I., & Li, J.-X. (2017). (pp. 181–223). Elsevier. https://doi.org/10.1016/B978-0-12-802447-8.00007-8
Abuchowski, A., van Es, T., Palczuk, N. C., & Davis, F. F. (1977). Journal of Biological Chemistry, 252(11), 3578–3581. https://doi.org/10.1016/S0021-9258(17)40291-2
Shi, D., Beasock, D., Fessler, A., Szebeni, J., Ljubimova, J. Y., Afonin, K. A., & Dobrovolskaia, M. A. (2022). Advanced Drug Delivery Reviews, 180, 114079. https://doi.org/10.1016/j.addr.2021.114079
Hoffman, A. S. (2016). Acta Biomaterialia, 40, 1–5. https://doi.org/10.1016/j.actbio.2016.05.029
Belliveau, N. M., Huft, J., Lin, P. J., Chen, S., Leung, A. K., Leaver, T. J., Wild, A. W., Lee, J. B., Taylor, R. J., Tam, Y. K., Hansen, C. L., & Cullis, P. R. (2012). Molecular Therapy - Nucleic Acids, 1, e37. https://doi.org/10.1038/mtna.2012.28
Mellinger, A., Lubitz, L. J., Gazaille, C., Leneweit, G., Bastiat, G., Lépinoux-Chambaud, C., & Eyer, J. (2023). International Journal of Pharmaceutics, 646, 123421. https://doi.org/10.1016/j.ijpharm.2023.123421
Hatakeyama, H., Akita, H., & Harashima, H. (2011). Advanced Drug Delivery Reviews, 63(3), 152–160. https://doi.org/10.1016/j.addr.2010.09.001
Richter, A. W., & Åkerblom, E. (1983). International Archives of Allergy and Immunology, 70(2), 124–131. https://doi.org/10.1159/000233309
Rosenblum, H. G., Gee, J., Liu, R., Marquez, P. L., Zhang, B., Strid, P., Abara, W. E., McNeil, M. M., Myers, T. R., Hause, A. M., Su, J. R., Markowitz, L. E., Shimabukuro, T. T., & Shay, D. K. (2022). The Lancet Infectious Diseases, 22(6), 802–812. https://doi.org/10.1016/S1473-3099(22)00054-8
Rosenblum, H. G., Gee, J., Liu, R., Marquez, P. L., Zhang, B., Strid, P., Abara, W. E., McNeil, M. M., Myers, T. R., Hause, A. M., Su, J. R., Markowitz, L. E., Shimabukuro, T. T., & Shay, D. K. (2022). The Lancet Infectious Diseases, 22(6), 802–812. https://doi.org/10.1016/S1473-3099(22)00054-8
Carreño, J. M., Singh, G., Tcheou, J., Srivastava, K., Gleason, C., Muramatsu, H., Desai, P., Aberg, J. A., Miller, R. L., study group, P., Pardi, N., Simon, V., & Krammer, F. (2022). Vaccine, 40(42), 6114–6124. https://doi.org/10.1016/j.vaccine.2022.08.024
Ju, Y., Lee, W. S., Pilkington, E. H., Kelly, H. G., Li, S., Selva, K. J., Wragg, K. M., Subbarao, K., Nguyen, T. H. O., Rowntree, L. C., Allen, L. F., Bond, K., Williamson, D. A., Truong, N. P., Plebanski, M., Kedzierska, K., Mahanty, S., Chung, A. W., Caruso, F., … Kent, S. J. (2022). ACS Nano, 16(8), 11769–11780. https://doi.org/10.1021/acsnano.2c04543
Guerrini, G., Gioria, S., Sauer, A. V., Lucchesi, S., Montagnani, F., Pastore, G., Ciabattini, A., Medaglini, D., & Calzolai, L. (2022). International Journal of Molecular Sciences, 23(16), 8838. https://doi.org/10.3390/ijms23168838
Bavli, Y., Chen, B.-M., Gross, G., Hershko, A., Turjeman, K., Roffler, S., & Barenholz, Y. (2023). Journal of Controlled Release, 354, 316–322. https://doi.org/10.1016/j.jconrel.2022.12.039
Alidjinou, E. K., Demaret, J., Corroyer-Simovic, B., Labreuche, J., Goffard, A., Trauet, J., Lupau, D., Miczek, S., Vuotto, F., Dendooven, A., Huvent-Grelle, D., Podvin, J., Dreuil, D., Faure, K., Deplanque, D., Bocket, L., Duhamel, A., Sobaszek, A., Hober, D., … Lefèvre, G. (2022). The Lancet Regional Health - Europe, 17, 100385. https://doi.org/10.1016/j.lanepe.2022.100385
Swadźba, J., Anyszek, T., Panek, A., Chojęta, A., Piotrowska-Mietelska, A., & Martin, E. (2023). Vaccines, 11(2), 278. https://doi.org/10.3390/vaccines11020278
Rosenblum, H. G., Gee, J., Liu, R., Marquez, P. L., Zhang, B., Strid, P., Abara, W. E., McNeil, M. M., Myers, T. R., Hause, A. M., Su, J. R., Markowitz, L. E., Shimabukuro, T. T., & Shay, D. K. (2022). The Lancet Infectious Diseases, 22(6), 802–812. https://doi.org/10.1016/S1473-3099(22)00054-8
Ishida, T., Harada, M., Wang, X. Y., Ichihara, M., Irimura, K., & Kiwada, H. (2005). Journal of Controlled Release, 105(3), 305–317. https://doi.org/10.1016/j.jconrel.2005.04.003
Suzuki, T., Suzuki, Y., Hihara, T., Kubara, K., Kondo, K., Hyodo, K., Yamazaki, K., Ishida, T., & Ishihara, H. (2020). International Journal of Pharmaceutics, 588, 119792. https://doi.org/10.1016/j.ijpharm.2020.119792
Wilson, S. C., Baryza, J. L., Reynolds, A. J., Bowman, K., Keegan, M. E., Standley, S. M., Gardner, N. P., Parmar, P., Agir, V. O., Yadav, S., Zunic, A., Vargeese, C., Lee, C. C., & Rajan, S. (2015). Molecular Pharmaceutics, 12(2), 386–392. https://doi.org/10.1021/mp500400k
Francia, V., Schiffelers, R. M., Cullis, P. R., & Witzigmann, D. (2020). Bioconjugate Chemistry, 31(9), 2046–2059. https://doi.org/10.1021/acs.bioconjchem.0c00366
Sherman, M. R., Williams, L. D., Sobczyk, M. A., Michaels, S. J., & Saifer, M. G. P. (2012). Bioconjugate Chemistry, 23(3), 485–499. https://doi.org/10.1021/bc200551b
Juang, V., Chang, C., Wang, C., Wang, H., & Lo, Y. (2019). Small, 15(49). https://doi.org/10.1002/smll.201903296
Hoang Thi, T. T., Pilkington, E. H., Nguyen, D. H., Lee, J. S., Park, K. D., & Truong, N. P. (2020). Polymers, 12(2), 298. https://doi.org/10.3390/polym12020298


